
Glycogen Storage
Glycogen is a carbohydrate or an extensively branched polymer of glucose stored primarily in the cells of the liver, skeletal muscle, and brain. A lower level is also found in plants (yeast and fungi) that do not contain chlorophyll. In animals, it is referred to as animal starch, which plays a significant role in nutrition. Due to more muscle mass, the quantity of glycogen in the muscle is about three times higher than that of the liver. Muscle glycogen is a fuel resource that supplies ATP during muscle contraction.
The primary function of liver glycogen is to maintain blood glucose levels during meals. Therefore, the liver glycogen storage increses during a well-fed state but decreases during fasting. However, glycogen storage disease happens due to abnormal storage of such molecules in the liver and skeletal muscle due to a lack of certain types of enzymes.
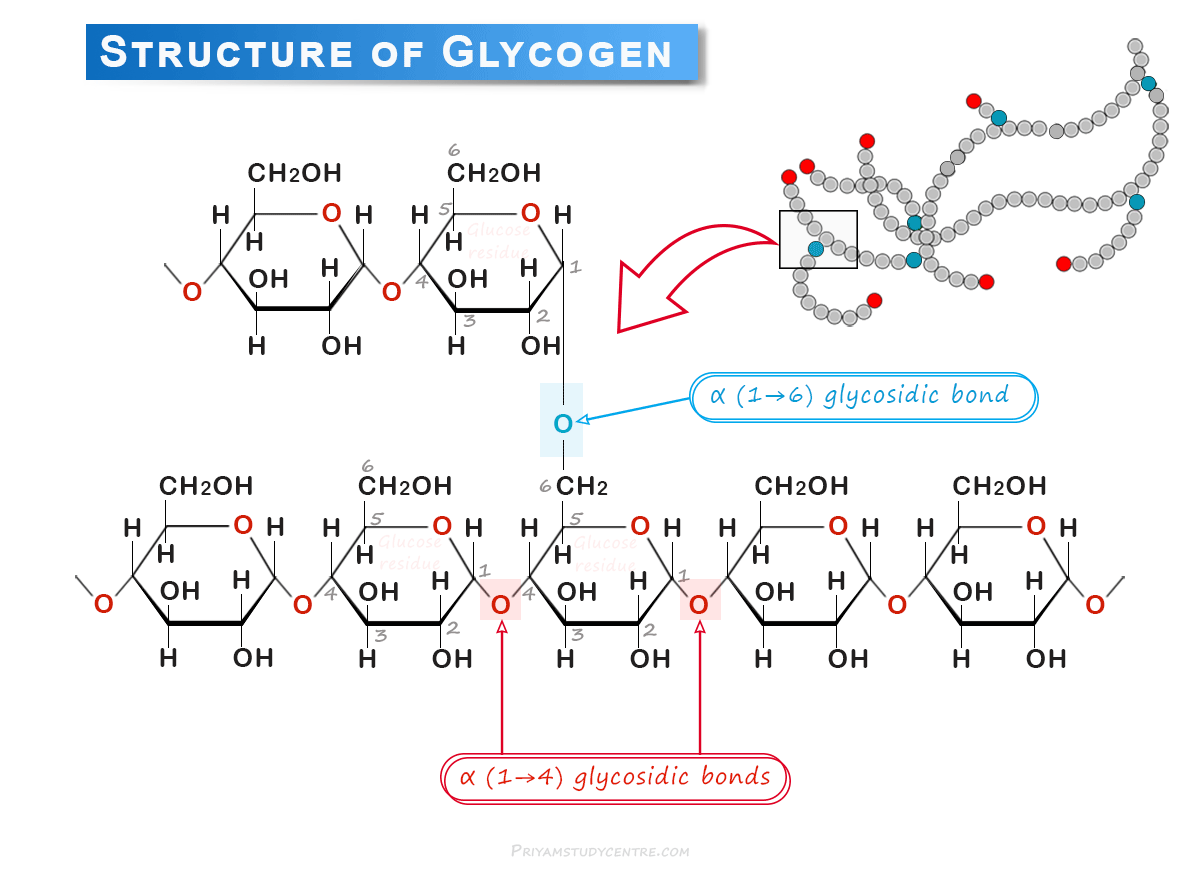
The glucose residues within this molecule are connected via two principal types of chemical bonding. Therefore, the structure of glycogen is similar to that of amylopectin, with more branches.
Glucose is the repeating unit in glycogen joined together by α (1→4) glycosidic bonds, and α (1→6) glycosidic bonds at branching points. The molecular weight and the number of glucose units in such molecules depend on the source from which it is obtained.
Glycogen Storage in the Human Body
Our body stores it mainly in the liver and skeletal muscle, and a small amount in our brain. However, our liver stores a greater ratio of glycogen than skeletal muscle, because our total muscle mass is greater than that of the liver.
The liver of an adult can store roughly 100–120 grams of glycogen, but the skeletal muscle of an adult stores roughly 400 grams. However, the weight of an adult is roughly 1.5 kg, but the skeletal muscle of an adult weighing 70 kg. Therefore, the concentration of glycogen in the liver is more than that of skeletal muscle.
Functions of Glycogen
Like starch, glycogen is the storage form of glucose molecules in animals or humans, and it is stored mostly in the liver and skeletal muscles. However, the quantity of storage glycogen in the muscle is about three times higher than that of the liver because the muscle in an animal has a higher mass than the liver.
It can be rapidly mobilized and generate energy in the absence of oxygen. On the other hand, fats or lipids are also stored in our bodies, but the metabolism of fat is very slow. It also needs oxygen for energy production. Therefore, fat may be considered as a fixed deposit, while glycogen is a current or savings account that is used at any time when energy is needed.
Liver Glycogen Storage
The primary function of liver glycogen is to maintain blood glucose levels during meals. It generally increases in a well-fed state, while it is depleted during fasting.
The glycogen store in the liver is mainly used to regulate blood glucose levels. Blood glucose is primarily regulated by the two hormones, insulin and glucagon.
During hypoglycemia or low glucose levels in our blood, our pancreas releases more glucagon. The enzyme glucagon can activate the glycogen store in our liver to convert it back to glucose, and it ener in our bloodstream.
The storage form of such molecules in the liver can also help in muscle activity and exercise. However, the stored glycogen in our muscles is the main source of energy for muscle activity.
Skeletal Muscle Glycogen Storage
The muscle glycogen can supply ATP during muscle contraction. Therefore, it is a fuel reserve for supplying ATP during muscle contraction.
Small amounts of glycogen are also stored in the other tissues and cells, such as the kidneys, red blood cells, white blood cells, and glial cells in the brain. Our brains depend on a continuous glucose supply that comes mostly from those molecules.
Synthesis of Glycogen
Glycogenesis is a process in which glucose can be synthesized from glycogen. It takes place in the cytosol and requires ATP and UTP, in addition to glucose.
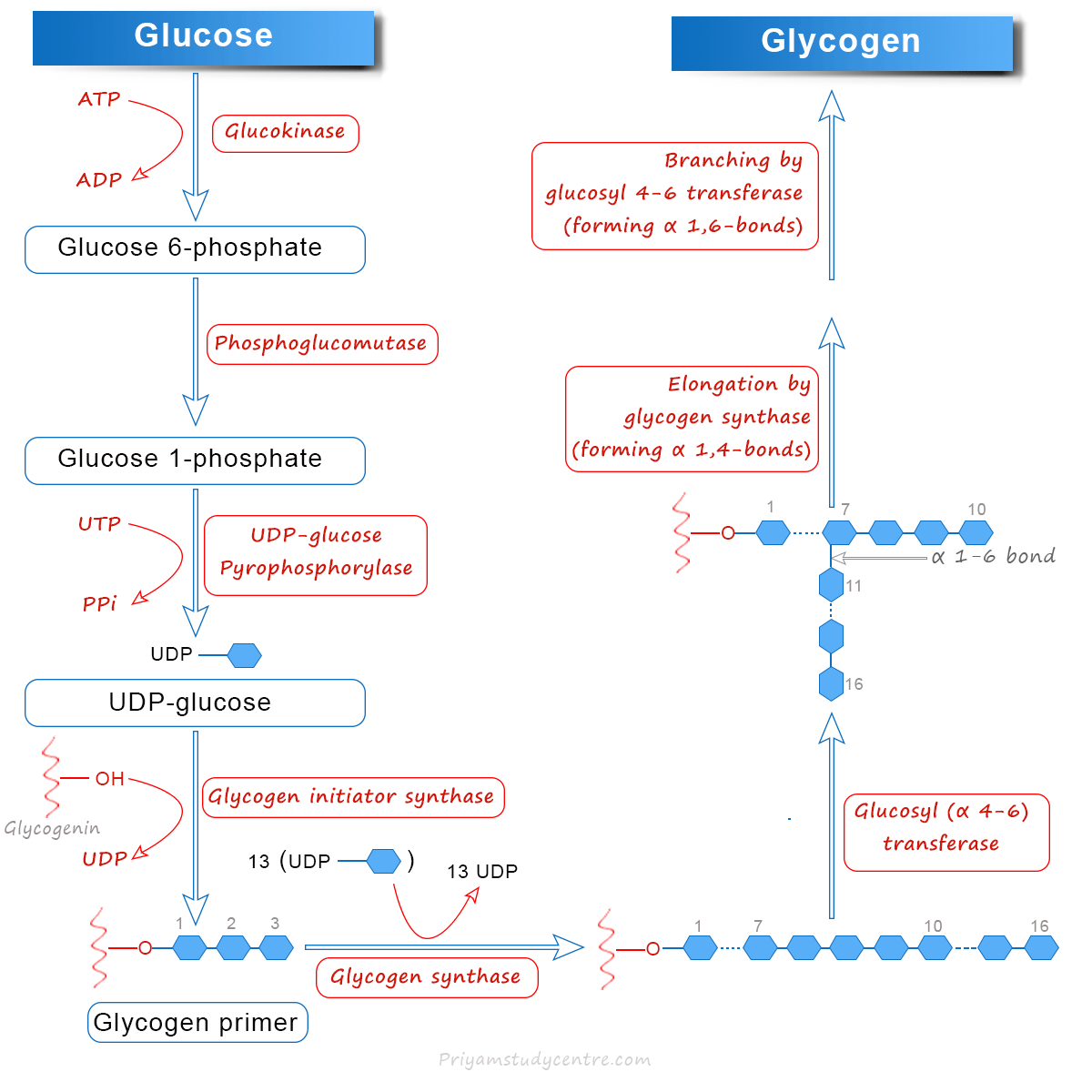
The overall biochemical reaction of such synthesis for the addition of each glucose residue is:
(glucose)n + Glucose + 2ATP → (Glucose)n+1 + 2ADP + Pi
Out of these two ATP, one is required for the phosphorylation of glucose, while the other is needed for the conversion of UDP to UTP.
Synthesis of UDP Glucose
The enzymes hexokinase in skeletal muscle and glucokinase in the liver can convert glucose to glucose 6-phosphate. Phosphoglucomutase catalyzes the conversion of glucose 6-phosphate to glucose 1-phosphate.
The enzyme UDP-glucose pyrophosphorylase can convert glucose 1-phosphate and UTP to uridine diphosphate glucose (UDPG).
Requirement of Primer to Initiate Glycogenesis
A small fragment of pre-existing glycogen must act as a primer to initiate the synthesis of glycogen.
Recently, we have seen that in the absence of glycogen primer, a specific protein (glycogenin) can accept glucose from UDPG. The hydroxyl group of the amino acid tyrosine of glyconenin is the site where the glucose molecule can be attached.
Generally, the first molecule of glucose is transferred to glycogenin by the enzyme glycogen initiator synthase. After that, glycogenin itself takes up a few glucose residues to form a fragment of primer. It commonly serves as an acceptor for the rest of the glucose molecules.
Glycogen Synthesis by Glycogen Synthase
1,4-glycosidic linkage in glycogen synthesis can be created by the enzyme glycogen synthase. Such an enzyme commonly transfers the glucose from UDP-glucose to the non-reducing end of glycogen to form α-1,4 linkages.
Formation of Branches in Glycogen
Glycogen synthase can catalyze the synthesis of a unbranched linear molecule with 1,4-α-glucosidic bonds. However, it formed a tree-like structure.
The branches are formed by the action of the branching enzyme, namely, glucosyl α-4, 6 transferase. Such enzymes transfer a small fragment of five to eight glucose residues from the non-reducing end of the glycogen chain by α-1, 4 linkages. It helps to form a new non-reducing end, besides the existing one.
Glycogenolysis
The degradation of stored glycogen in the liver and skeletal muscle constitutes glycogenolysis. However, the pathways for the synthesis or storage, and degradation of glycogen are not reversible. It is commonly degraded by the breaking of α-1,4- and α-1,6-glycosidic bonds.
Glycogen Phosphorylase
The enzyme glycogen phosphorylase can cleave the α-1,4-glycosidic bonds from non-reducing ends to yield glucose 1-phosphate.
Such a process continues until four glucose units remain on either side of the branching point, and it is called phosphorolysis.
Action of Debrancing Enzyme
A bifunctional enzyme or debranching enzyme that has two enzyme activities present on a single polypeptide can cleave the branches of glycogen. It removes a fragment of three or four glucose residues attached at the branch and transfers them to another chain. Therefore, one α-1,4-bond is cleaved, and the same α-1,4-bond is made, but the places are different.
The enzyme amylo α-1,6-glucosidase can break the α-1,6-bond at the branch with a single glucose residue or a free glucose molecule. The remaining molecule is again available for the action of phosphorylase and debranching enzyme to repeat a similar biochemical reaction.
Formation of Glucose 6-phosphate and Glucose
Glucose 1-phosphate and free glucose are produced by the combined action of phosphorylase and debranching enzyme. Then the enzyme phosphoglucomutase can convert glucose 1-phosphate to glucose 6-phosphate. The conversion of glucose 6-phosphate to glucose may depend on the glycogen storage tissues.
The liver, kidney, and intestine contain the enzyme glucose 6-phosphatase, and it cleaves glucose 6-phosphate to free glucose molecules. However, such an enzyme is absent in muscle and brain, hence free glucose can not be produced in such tissues. Therefore, the liver is the major glycogen storage organ that provides glucose for various biochemical reactions.
Glycogen Synthesis and Degradation
Glycogen synthesis and degradation are essential for maintaining the blood glucose level in the human body. Such synthesis and degradation processes are generally controlled by the enzymes glycogen synthase and glycogen phosphorylase. The regulation of these enzymes is carried out by the following mechanism:
- Allostretic Regulation
- Hormonal Regulation
- Influence of Calcium
Allosteric Regulation of Glycogen
The activities of glycogen synthase and phosphorylase are allosterically regulated by certain metabolites. Generally, the synthesis of glycogen increses when substrate availability and energy level are high. Similarly, the glycogen breakdown is enhanced when glucose concentration and energy level are low.
In a well-fed state, the availability of glucose 6-phosphate is high, which allosterically activates the enzyme synthase for a larger amount of synthesis. On the other hand, glucose 6-phosphate and ATP allosterically inhibit the enzyme phosphorylase. Free glucose is also an inhibitor that inhibits the activities of phosphorylase.
Hormonal Regulation of Glycogen
Through a complex series of biochemical reactions, many hormones may regulate the synthesis and degradation of glycogen molecules.
Hormones like epinephrine, norepinephrine, and glucagon in the liver activate adenylate cyclase to increase the production of cAMP. Similarly, the hormone insulin increses the activity of phosphodiesterase in the liver and breaks down cAMP.
The process of glycogenesis may be regulated by the enzyme synthase that exists in two forms, a form and b form. Synthase a form is the most active, but Synthase b form is an inactive form of such an enzyme. Synthase a form can be converted to synthase b form by a cAMP-dependent protein kinase.
Effect of Ca+2 Ions
During muscle contraction, Ca+2 ions can be released from the sarcoplasmic reticulum. Calcium ions bind to calmodulin-calcium modulating protein, and directly activate phosphorylase kinase without the involvement of cAMP-dependent protein kinase.
Generally, an elevated glucagon or epinephrine level increses glycogen degradation, whereas an elevated insulin level results in increses glycogen synthesis.
Glycogen Storage Disease (GSD)
Glycogen storage disease (GSD) or glycogenoses is a rare type of inherited metabolic disorder where a person is born without certain enzymes that help to make and break down glycogen. Therefore, such types of health diseases generally happen due to abnormal storage of glycogen in the liver and skeletal muscle due to a lack of certain types of enzymes. The overall incidence found is around 1 per 40,000 births.
The inability to form glucose from glycogen can lead to long-term complications such as hypoglycemia and exercise-induced weakness in the human body.
Glycogen storage disease 0 occurs due to the deficiency of the enzyme glycogen synthase in the liver. However, it does not store extra molecules in the liver, but it has various symptoms comparable to those of other GSDs.
The incidence of type I glycogen storage disease, or von Gierke’s disease found in 1 per 200,000 persons and is transmitted by an autosomal recessive trait. Such a type of health disease in the human body can create the following types of biochemical disorders:
- Fasting hypoglycemia
- Lactic acidemia
- Hyperlipidemia
- Hyperuricemia







