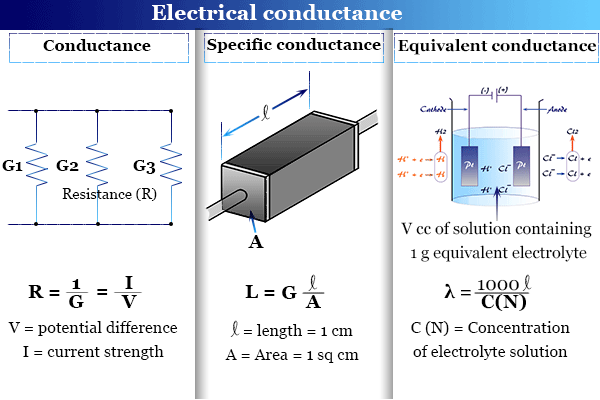
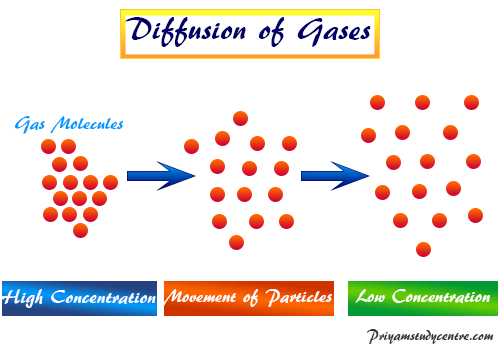
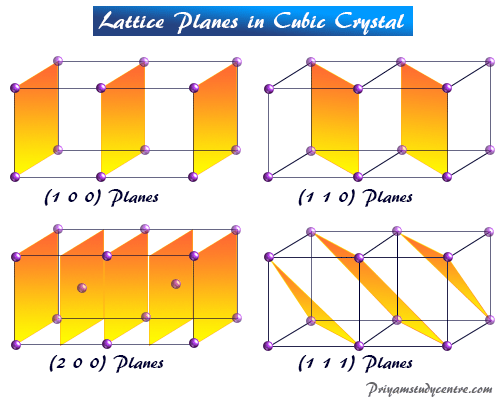
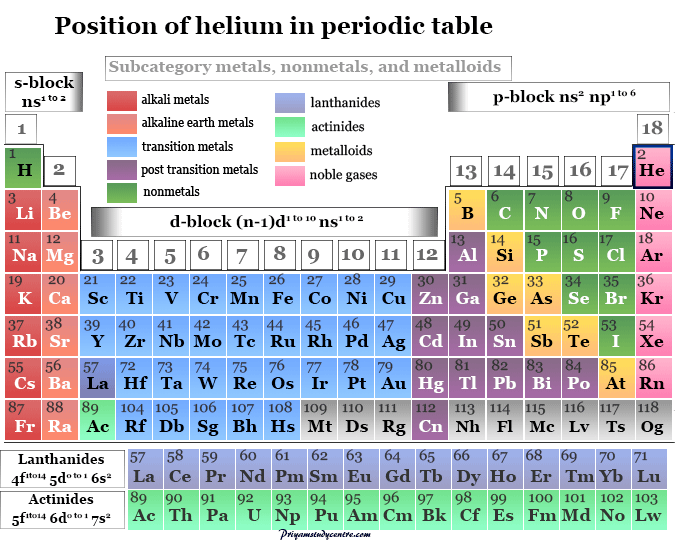
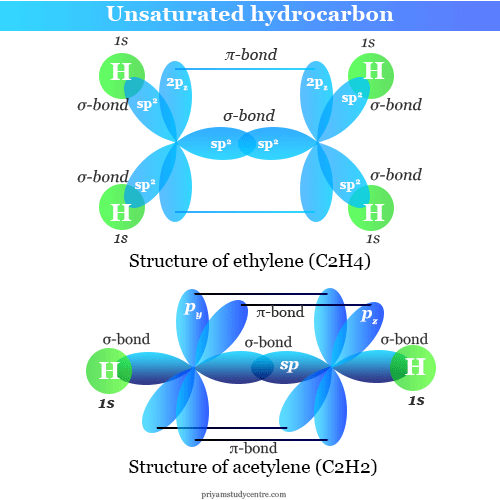
Learning Chemistry Online
Learning chemistry online free chemistry courses in PSC guides and helps by sharing articles or topics for general, medical, environmental science, and chemical engineering beginners. In online learning chemistry, our basic aim is to share articles or topics related to physical, inorganic, organic chemistry, analytical methods or techniques, spectroscopy, biochemistry, medicinal, and environmental science to guide or help medical and engineering students and researchers in their studies. In our online free chemistry courses, we constantly add or update new articles on our website for students who want to learn or study from home.
Online Free Chemistry Courses
Online free chemistry courses in PSC not only share the definition, formula, principles, methods, and techniques of chemical science. It also guides or helps you to better learning from home.
In free online chemistry courses, we also provide quizzes and frequently asked questions (FAQ) with their answers. The free online quizzes on chemistry articles or topics help general, medical, and chemical engineering beginners to test their knowledge for upcoming exams.